Poster Session 2025
- Home
- Amanda N. D. Adams
- Scarlet Au
- Dayakar Badri
- Alexander Chan
- Marina Chen
- Jose Collado
- Deepika Dinesh
- Danyue Dong
- Jiayi Duan
- Guilherme Fahur Bottino
- Jasmine Garcia
- McKenzie Gehris
- Ishika Gupta
- Mariss Haddad
- Anna Happel
- Kayla Hazlett
- Lauren Hutchinson
- Jordan Jensen
- Charles Jo
- María Alejandra Jové
- Tanya Karagiannis
- Younhun Kim
- Jae Sun Kim
- Helle Krogh Pedersen
- Valeria Lugo-Mesa
- Wenjie Ma
- Daniel MacDonald
- Sithija Manage
- Olivia Maurer
- Nicholas Medearis
- Steven Medina
- Maeva Metz
- Xochitl Morgan
- Jacob Nearing
- William Nickols
- Etienne Nzabarushimana
- Askarbek Orakov
- Mustafa Özçam
- Tathabbai Pakalapati
- Audrey Randall
- Yesica Daniela Roa Pinilla
- María Alejandra Rodriguez-Alfonso
- Patrick Rynkiewicz
- Laura Schell
- Jiaxian Shen
- Meghan Short
- Wilhelm Sjöland
- Daniel Sprockett
- Melissa Tran
- Benjamin Tully
- Chahat Upreti
- Akshaya Vasudevan
- Emily Venable
- Jasmine Walsh
- Dongyu Wang
- Kai Wang
- Ya Wang
- Zhongjie Wang
- Yilun Wu
- Ji Youn Yoo
Poster Session 2025
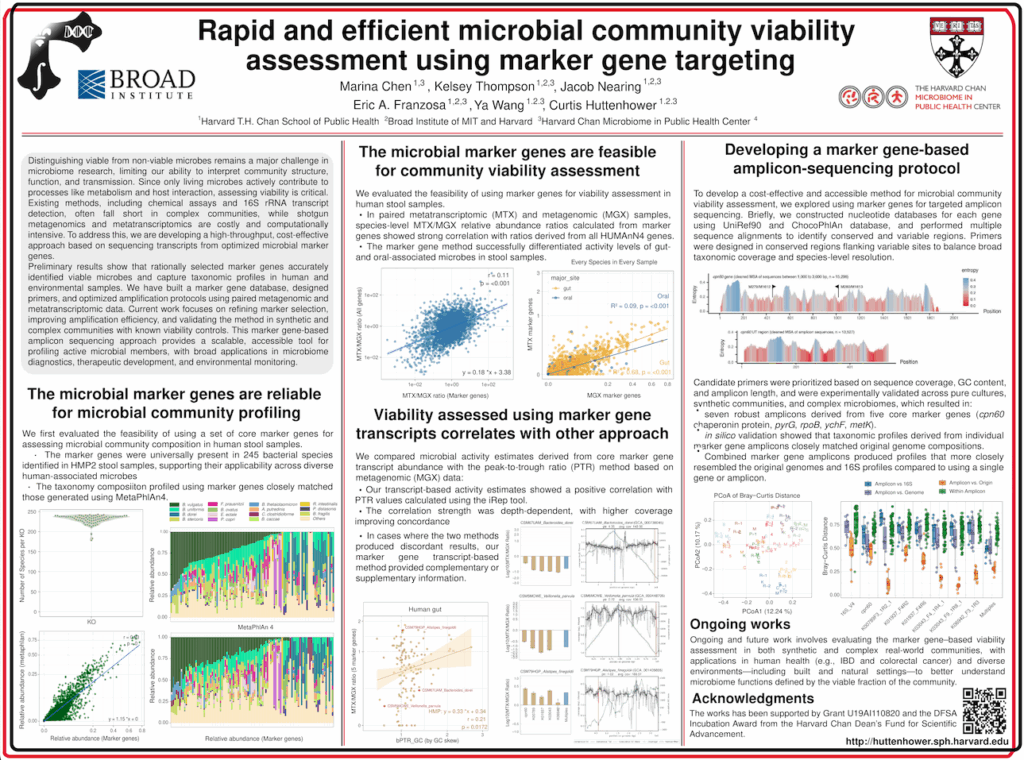
Rapid and efficient microbial community viability assessment using marker gene targeting
Presented By: Ya Wang
Distinguishing viable from non-viable microbes remains a major hurdle in microbiome research, limiting our ability to fully understand microbial community structure, function, and transmission. Microbial viability is critical for interpreting the biological and ecological roles of microbiomes, as only living microbes actively participate in processes such as metabolism, signaling, and host interactions. Existing methods, such as chemical-based viability assays and 16S rRNA transcript detection have proven sub-optimal in complex communities, while shotgun metagenomics and metatranscriptomics, though informative, are often prohibitively expensive and computationally demanding. To address these limitations, we are developing a novel, high-throughput viability assessment approach based on the sequencing of optimized marker gene transcripts.
Our method combines rational marker gene selection, in silico primer design, and amplicon sequencing to enable precise, scalable differentiation of actively transcribing microbes within diverse communities. Preliminary work shows that rationally selected marker genes can effectively distinguish viable microbes and provide accurate taxonomic and functional profiles in human and built environment samples. We have constructed a marker gene database, designed and evaluated primers, and developed amplification protocols based on paired metagenomic and metatranscriptomic datasets. Moving forward, we are refining marker gene amplicon design, optimizing amplification protocols for cost-effectiveness and accuracy, and validating the approach in both synthetic communities and complex natural samples with known viability controls. Once established, this workflow will offer an accessible, culture-independent solution for microbiome viability profiling. This approach has broad implications for advancing microbiome-based diagnostics, therapeutic development, and environmental surveillance by providing a much-needed tool to quantify the active, functional members of microbial communities.